

Although we are interested in problems across the spectrum of materials science, our current research is mainly focused on three areas: quantum technology, green energy generation, and method development.
We are always very interested to hear from anyone who would be interested in working with us on any of these research areas, whether that is as a PhD student, a postdoc, a research fellow, or an established academic. If you would also be interested in working with us, please take a look at the list of opportunities here, or get in contact with Joe.
Materials for quantum technology

The development of new and exciting quantum technologies has the potential to change the world. Perhaps the most well-known of these technologies is quantum computing, but there are many potential applications for quantum sensing, quantum communication, and many others.
An important part of developing these technologies is finding and understanding the materials that they will be made from. First principles modelling has an important role to play in this, as it gives access to atomic-scale understanding of the quantum mechanical effects that are vital to these technologies.
Our work in this area currently largely focuses on understanding the behaviour of point defects in crystalline materials, sometimes known as colour centres, some of which are excellent candidates for quantum technology applications. The most famous of these is perhaps the nitrogen-vacancy centre in diamond, but there are a large number of possible defect centres and host materials with promising properties. We are also interested in molecular systems with more exotic excited state properties, with applications in quantum technology. We use simulation to drive understanding of these systems in collaboration with experimental groups, identifying unknown defects, understanding their interactions with their environment, and suggesting new pathways for research.
Collaborators
- Prof Jason Smith (Oxford)
- Prof Hannah Stern (Oxford)
- Prof Sascha Feldmann (EPFL)
General project areas
- Understanding interactions between common defects and colour centres in materials for quantum technology
- Understanding interface effects on colour centres in materials for quantum technology
- Modelling perturbations to colour centres in two-dimensional candidate materials for quantum technology
- Controlling interplay between optical absorption and circular dichroism in large-scale systems
Materials for green energy generation
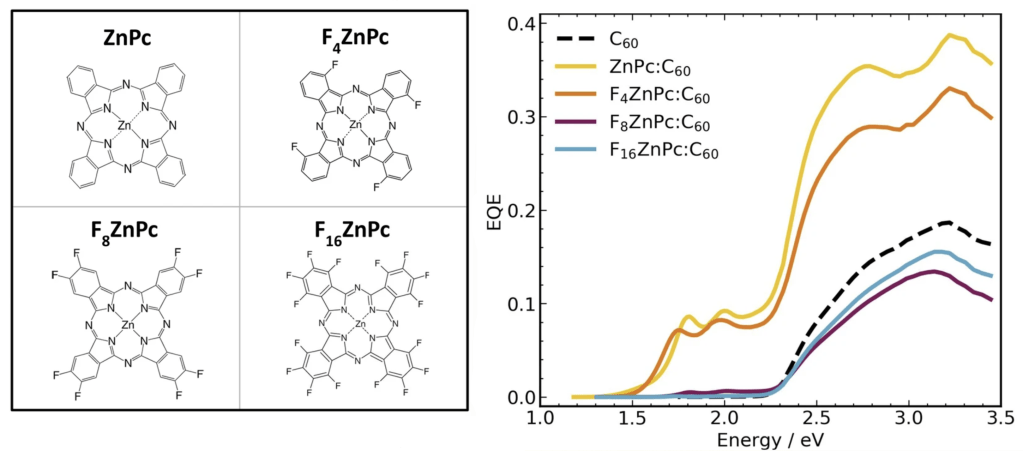
Satisfying the world’s energy demands without further worsening the climate crisis is one of the biggest challenges in modern science and technology. A key component of this is solar energy. Silicon solar cells currently dominate the market, but there are several emerging photovoltaic materials that promise low-cost, high-efficiency photovoltaics with other desirable properties, such as flexibility or low weight.
The closest of these emerging materials to commercialisation are organic photovoltaics. These are made up of organic molecules, consisting of either long-chain polymers or smaller molecules, often with multiple molecular species mixed together. Although the former class of materials is generally more developed, the latter class is easier to fabricate. Both classes typically have highly disordered structures.
Our work in this area currently focuses on understanding the role of disorder in modifying the excited state properties of small molecule photovoltaics, using cutting edge first principles techniques that are capable of modelling these highly disordered systems. Interactions between molecules can significantly shift their light absorption properties, and the disorder in these systems means the molecules can be exist in a wide array of different environments. If we understand how these environments affect the properties of the molecules, using both simulation and experiment, we can use this information to design better photovoltaics, through adjusting the mixture of molecules, the fabrication process, and other methods.
Collaborators
- Prof Moritz Riede (Oxford)
General project areas
- Unravelling the impact of disorder on optical absorption properties of small-molecule organic photovoltaics
Materials modelling method development

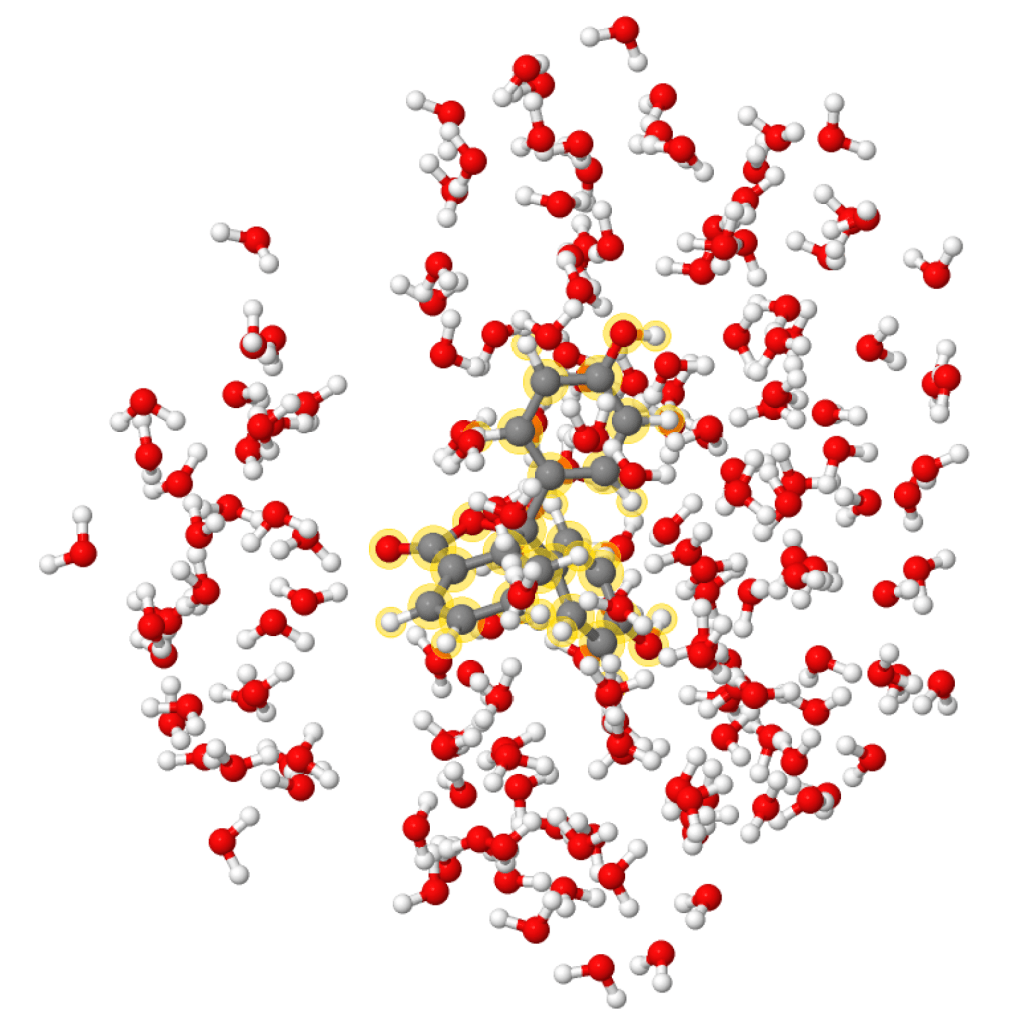
Density functional theory (DFT) is one of the most popular first principles materials modelling methods, thanks to its combination of simplicity, computational efficiency, and ability to accurately predict many properties of many materials. These properties have contributed to its domination of the field of computational materials modelling, and the flourishing of many DFT-based codes, including CASTEP and ONETEP, two codes developed in the Prentice Group.
However, there are several well-known issues with the standard formulation of DFT and its derivatives such as time-dependent DFT (TDDFT) that can restrict its usage for more complex problems, such as cubic scaling, underestimation of the band gap, and incorrect treatment of charge transfer. More advanced techniques, such as linear-scaling DFT or the GW approximation, that can deal with one or more of these issues are available, but often significantly increase the computational cost.
Our work in this area therefore focuses on bringing these higher-level methods to bear on complex systems by making them more computationally efficient and applicable. We mainly focus on developing quantum embedding techniques, particularly linked to linear-scaling DFT; however, we also work on other DFT-based techniques for modelling both electronic and vibrational excitations, including vibrational spectroscopy and X-ray absorption. Much of this development takes place within the ONETEP and CASTEP codes, both of which are UK flagship DFT codes, bringing our work to a wide-spread audience in academia and industry.
Collaborators
- Prof Arash Mostofi (Imperial)
- Prof Peter Haynes (Imperial)
- Prof Chris Skylaris (Southampton)
- Dr Jacek Dziedzic (Southampton/Gdansk)
- Prof Nick Hine (Warwick)
General project areas
- Quantum embedding of GW and Bethe-Salpeter equation methods in linear-scaling DFT: accurate excitons in large-scale heterogeneous systems
- Quantum embedding of wavefunction-based methods in linear-scaling DFT: chemical accuracy in large-scale heterogeneous systems
- Simulating L-edge X-ray absorption from first principles in the solid state
Other interests
We also use first principles modelling to understand the electronic structure of unconventional iron-based superconductors, working closely with experimental groups to match our computational results against experiment. By understanding how external factors such as strain, chemical substitution, and magnetism affect the electronic structure computationally, we can hopefully begin to understand how superconductivity emerges in such systems.
Collaborators
- Prof Amalia Coldea (Oxford)
